Capturing hydrogen transfer from modeling: a small step towards simulating chemical reactions by computers
All the matter of the world around us are made of atoms that are joined to form molecules. Chemists can create new molecules by changing connectivity of atoms in molecules through chemical reactions. Researchers can control reaction pathway to generate expected molecules with superior functions after uncovering reaction magic. To explore reaction mechanisms underneath experiments, theoretical models have been developed. By leveraging the computers, researchers can simulate reaction processes using theoretical approaches besides the traditional experiment techniques in lab. People begin to walk deep into the atomic world, which greatly facilitate chemical synthesis and material design. Due to the growing success achieved by computational modeling, Nobel Prize was awarded to three chemist “for the development of multiscale models for complex chemical systems” in 2013. However, the early version of multiscale models, for example, the hybrid quantum mechanical and molecular mechanical (QM/MM) method, cannot reach the timescale of reaction occurrence. To overcome this issue, the multiscale modeling method is combined with enhanced sampling method to realize sampling within limited simulation timescale. The first try is demonstrated by implementing QM/MM potential with the selective integrated tempering sampling (SITS) method.
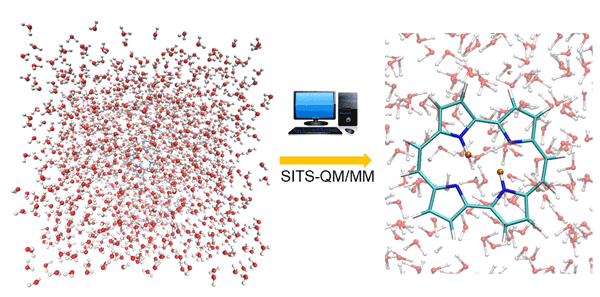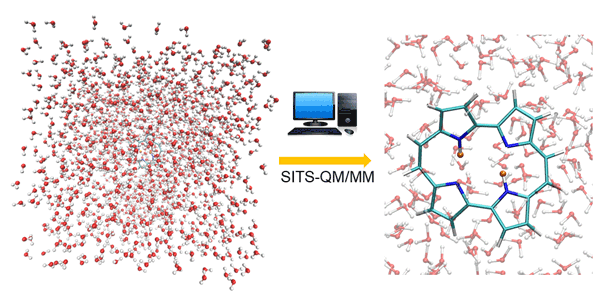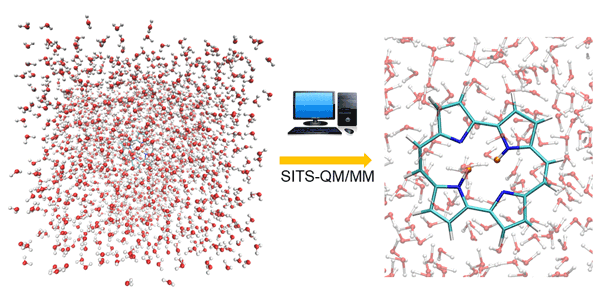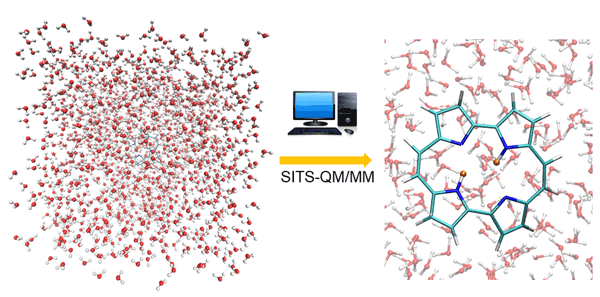
Fig. 1. Double proton transfer process in porphycene simulated by SITS-QM/MM method. The protons are highlighted with orange color.
The combined SITS-QM/MM is proposed to investigate the chemical reactions in solution. The applicability is validated by using two representative double proton (hydrogen atom with positive charge) transfer reactions, independent proton transfer in 3-3’-bis-malondialdehyde (MDH) and correlated proton transfer processes in porphycene (PPC). SITS-QM/MM molecular dynamics simulations can monitor in real time distance changes of hydrogen bond as illustrated in Figure 1, which is beyond the resolution of experimental techniques. The time delay between two consecutive proton transfers serves as important clues to distinguish independent and correlated process. To obtain more insight, the free energy barrier that is required for hydrogen bond breakage is computed along the reaction pathway, which cannot be captured by normal molecular dynamics simulations. One noticeable finding is that the existence parallel reaction pathway can lead to the pseudo free energy change profile. The conditional distance distribution and free energy profile have been well established to identify independent and correlated proton transfer processes for different molecules. The combination of SITS and QM/MM make it possible to identify possible reaction paths, intermediate and product states without prior knowledge of reaction mechanisms. Moreover, the hydrogen/deuterium (H/D) isotope effects on the double proton transfer in PPC are also investigated in the later study. The structural analysis and the free energy shifts of double proton transfer process indicate that the asymmetric isotopic substitution in PPC compress the covalent hydrogen bonds and alter the location of original transition state of unsubstituted PPC. SITS-QM/MM can provide free energy barriers and structural changes during reaction, which provides chemical insights for the design of chemical reaction pathway. The protocol can be easily extended to explore and optimize potential halogen or deuterium substitution of the hit discovery and lead optimization stages of drug design, which would be beneficial for both academic studies and industry applications.
The multiscale molecular modeling combined with enhanced sampling provides an initial trial for the road map of the capturing chemical reaction steps in real-time. We are excited to look forward to more advanced theoretical methods to simulate more complex reactions by computers in the future.
Liangxu Xie
Institute of Bioinformatics and Medical Engineering, School of Electrical and Information Engineering, Jiangsu University of Technology, China
Publication
Enhanced QM/MM sampling for free energy calculation of chemical reactions: A case study of double proton transfer
Liangxu Xie, Huimin Cheng, Dong Fang, Zhe-Ning Chen, Mingjun Yang
J Chem Phys. 2019 Jan 28

Related Articles:
 | UCLA researchers develop high-sensitivity… A significant advancement for point-of-care medical diagnostics, a team of researchers from UCLA has introduced a deep learning-enhanced, paper-based vertical flow assay (VFA) capable of detecting cardiac troponin I (cTnI)… |
 | UCLA researchers pioneer AI-based tissue staining to… Los Angeles, CA – September 10, 2024 – Researchers at the University of California, Los Angeles (UCLA) have pioneered a groundbreaking approach in the imaging and detection of amyloid deposits… |
 | Improving assessment of arthritis models to better… Rheumatoid Arthritis (RA) is a common inflammatory disease that is characterized by swelling and tenderness of multiple joints. The resulting pain and joint stiffness cause disability for patients and treatment… |
 | Unlocking new treatments for bone diseases: using… Despite appearances, bones are a constantly changing organ in the body. Cells that produce new bone (named osteoblasts) and cells that break down bone (named osteoclasts) work in harmony to… |
 | Making Christmas trees under duress, or how cells… Some of the most enduring images for a molecular biologist are electron microscopy micrographs of the so-called “Christmas trees”, famously first observed by Oscar Miller from newt oocytes in 1969.… |
 | Waking a sleeping giant: How increasing Africa's… Africa is home to 1.4 billion people, of whom 55 million suffer from cardiovascular disease (CVD). Despite this, Africa contributes to only 1.7% of the world’s cardiovascular science. We studied… |





Leave a Reply
You must be logged in to post a comment.